Článek
Bohužel, situace popsaná výše není nijak výjimečná. Když se mluví o dědičných onemocněních, mnoho z nás si v duchu oddychne – v naší rodině se přece nic takového nevyskytuje. Jenže tahle představa má zřejmou, zásadní slabinu: v každé rodině musí jednou existovat první případ, u kterého se nemoc začne zřetelně projevovat.
U chorob, jako je Huntingtonova nemoc, se tento mechanismus ukazuje obzvlášť názorně. Nejde totiž jen o to, zda je genetická změna přítomná, ale také o její rozsah, protože až od určité hodnoty vede k nemoci. Jinými slovy, po mnoho generací může být chyba malá na to, aby nemoc způsobila, ovšem při každém přenosu na potomka se může dále zvětšit, až přesáhne onu kritickou hranici. Ale o tom blíže až za chvíli.
Co je Huntingtonova nemoc?
Jde o dědičné onemocnění mozku, při kterém postupně zanikají neurony, především v oblastech zodpovědných za řízení pohybu, myšlení a emocí. Patří mezi neurodegenerativní choroby, což znamená, že se stav pacienta v čase pomalu, ale trvale zhoršuje.
Nemoc se nejčastěji začne projevovat v dospělosti, typicky mezi 30. a 50. rokem života (jak ale uvidíme za chvíli, to závisí i na rozsahu dané genetické změny). Právě tento pozdní nástup má ale zásadní důsledky – mnoho lidí už v té době žije běžný život, má práci, partnera a často i vlastní děti, aniž by tušili, že v sobě nesou genetickou změnu.
Průběh bývá pozvolný, ale zasahuje několik základních oblastí fungování člověka. Objevují se mimovolní, trhavé pohyby těla, označované jako chorea. Tyto pohyby mohou působit dojmem neklidného „tančení“, nejsou rytmické ani vědomě ovladatelné, ale přesto mohou připomínat jakési neuspořádané taneční kroky. Právě odtud pochází i historické označení tanec svatého Víta, které se dříve používalo pro různé stavy spojené s podobnými pohybovými projevy. Postupně se pak kontrola nad svaly a koordinace zhoršuje.
Současně dochází ke změnám v myšlení, například k potížím s pamětí, soustředěním nebo plánováním. Tyto kognitivní potíže se prohlubují a u většiny pacientů přecházejí do obrazu demence, která výrazně omezuje schopnost samostatného fungování. Neméně důležité jsou i změny psychické, jako jsou deprese, úzkosti, podrážděnost nebo změny osobnosti. Průměrná doba přežití je kolem 15 let.
Z pohledu neuropatologie jsou nejvýrazněji postižena hluboká mozková jádra, zejména striatum, přičemž postupně dochází i k postižení mozkové kůry, což je dobře vidět na obrázku níže.
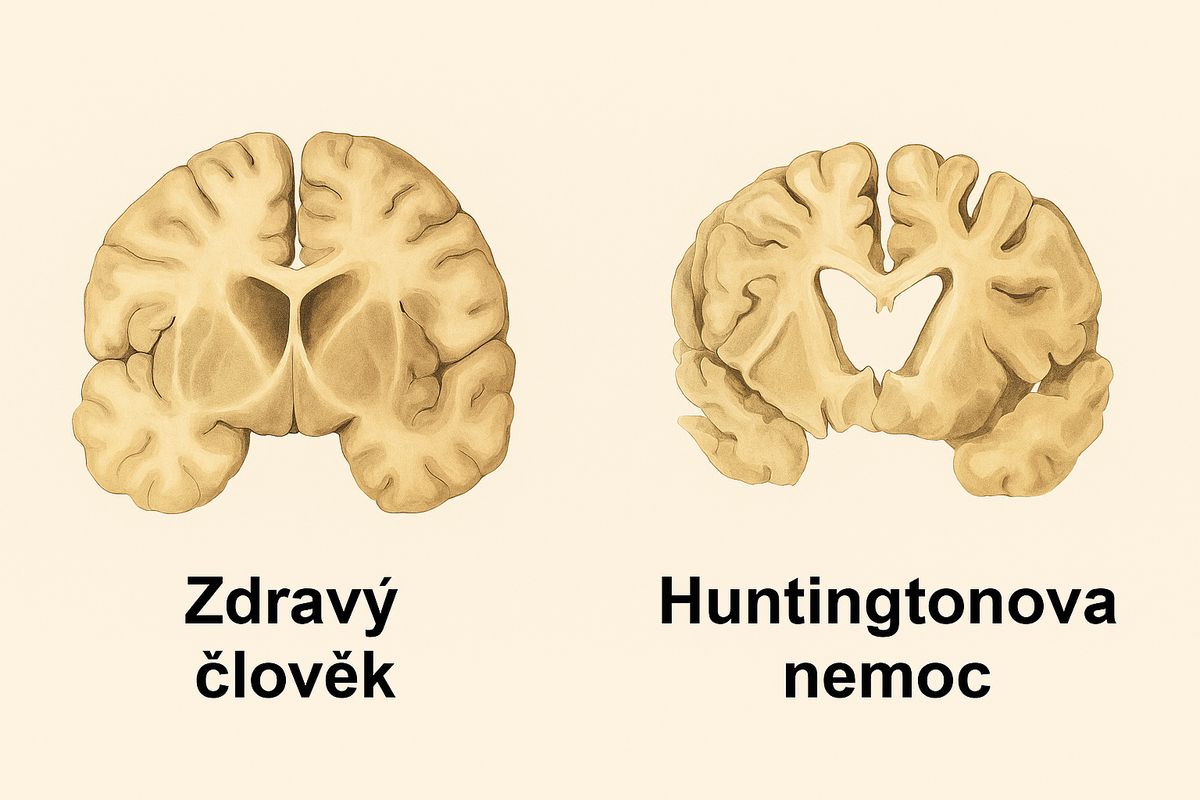
Srovnání řezů mozkem zdravého člověka a pacienta s pokročilou Huntingtonovou nemocí.
Proč nemoc vzniká aneb co se děje v DNA
Příčina Huntingtonovy nemoci je dnes popsána velmi přesně, a zároveň představuje jeden z učebnicových příkladů toho, jak může relativně drobná změna v DNA vést k zásadním biologickým důsledkům. Abychom mohli tuto změnu pochopit, je potřeba začít úplně od základní stavební úrovně.
DNA je zapsána pomocí čtyř základních „písmen“ – A (adenin), C (cytosin), G (guanin) a T (thymin). Jednotlivá písmena ale nemají význam sama o sobě, ten vzniká teprve tehdy, když se spojí do trojic (tripletů). Každá taková trojice funguje jako jednoduchý pokyn: říká buňce, jakou stavební jednotku má použít při tvorbě bílkoviny. Celý genetický kód tak lze chápat jako dlouhý text, kde slova nemají různou délku, ale vždy přesně tři písmena.
Jednou z takových trojic je CAG (cytosin–adenin–guanin). Tato kombinace se běžně vyskytuje v DNA každého člověka a sama o sobě není nijak škodlivá. V jednom konkrétním genu se však CAG opakovaně řadí za sebou, běžně v počtu nižším než 27.
Každá trojice CAG nese informaci pro vznik jedné konkrétní aminokyseliny, zvané glutamin. Pokud se tedy CAG v genu opakuje například dvacetkrát, vznikne při tvorbě bílkoviny úsek složený z dvaceti glutaminů za sebou, který funguje normálně.
Rozhodující je ale okamžik, kdy se počet těchto opakování dostane nad běžnou hodnotu. Čím více se trojice CAG v genu opakuje, tím delší glutaminový řetězec vzniká – a právě toto zvýšení počtu opakování (nad hraniční hodnotu) je přímou příčinou Huntingtonovy nemoci.
Při nadměrném prodloužení se bílkovina začne špatně skládat, ztrácí svou původní funkci a zároveň získává vlastnosti, které jsou pro nervové buňky škodlivé. Nejde tedy o cizí bílkovinu, ale o normální bílkovinu s nepřiměřeně prodlouženou částí, která postupně narušuje fungování mozkových buněk.
Proč tedy může mít zdravý rodič nemocné děti?
Jak jsem popsal na případu v úvodu, jedna z nejzáludnějších vlastností Huntingtonovy nemoci je skutečnost, že se může objevit u dítěte rodiče, který byl celý život zdravý a neměl žádné příznaky, což pro rodiny o často působí jako naprostý šok.
Vysvětlení znovu souvisí s opakováními CAG. Jak už víme, menší počet těchto opakování je normální a nezpůsobuje žádné potíže. Existují ale lidé, kteří mají počet opakování na horní hranici normy. Sami nikdy neonemocní, ale jejich DNA je v této části méně stabilní.
A zde se dostáváme ke klíčovému problému. Při přenosu genetické informace z rodiče na dítě se totiž může stát, že se počet CAG opakování mírně změní, nejčastěji se nepatrně zvýší (i když teoreticky může i klesnout). Rodič přitom zůstává zdravý, ale dítě už může zdědit delší variantu, která se posune do rizikového pásma.
To má velmi konkrétní důsledky. Pokud se dítě dostane do rozmezí 36–39 opakování, nachází se v šedé zóně: nemoc se u něj může, ale nemusí během života projevit. Jakmile je však počet opakování 40 a více, riziko rozvoje Huntingtonovy nemoci je prakticky jisté, přičemž s vyšším počtem opakování klesá věk nástupu nemoci. Přehled těchto hodnot a jejich významu je shrnut v tabulce níže.
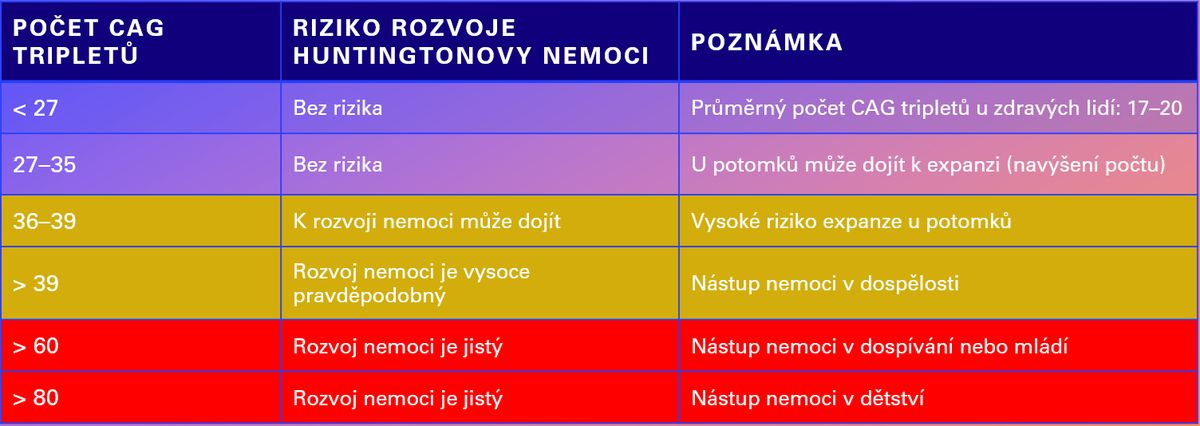
Přehled pásem hodnot CAG tripletů a riziko rozvoje nemoci.
Právě tímto mechanismem může vzniknout situace, kdy se Huntingtonova nemoc v rodině objeví zdánlivě poprvé. Ve skutečnosti tam genetická změna byla přítomná už dříve, ale dlouho zůstávala pod hranicí, která by vedla k onemocnění. Až v jedné generaci dojde k nenápadnému prodloužení CAG sekvence natolik, že je tento práh překročen.
Důležité je také to, že toto prodlužování se častěji děje při přenosu od otce. Dědičná informace se u mužů během života mnohokrát kopíruje při tvorbě spermií a právě při tomto opakovaném přepisování má opakující se úsek CAG větší sklon se ještě prodloužit. Proto se Huntingtonova nemoc v praxi častěji objeví v linii přes otce než přes matku.
To má velmi konkrétní důsledky: z pohledu jedné generace může být nemoc náhlá a nečekaná, z pohledu biologie je ale výsledkem postupného hromadění změny, která byla dlouho pod prahem onemocnění.
Genetické testování
Testování na Huntingtonovu chorobu u nás zahrnuje několik přístupů podle důvodu vyšetření. Provádí se prediktivní testování u osob bez příznaků, které jsou ale v riziku kvůli rodinné anamnéze, dále diagnostické testování u pacientů s podezřením na onemocnění a také prenatální či preimplantační testování, které může mutace odhalit.
U rizikových osob je celý proces vždy doprovázen odborným psychologickým a psychiatrickým poradenstvím a více konzultacemi před i po testu. Důvodem je, že výsledek může mít zásadní psychický dopad a ovlivnit nejen testovaného, ale i jeho rodinu, proto se rozhodnutí o testování pečlivě zvažuje v rámci podpory odborníků.
Jen na okraj - možná si pamatujete na seriál Dr. House (House, M.D.), kde se tato problematika objevila velmi názorně. Jedna z hlavních postav zde pocházela z rodiny zatížené Huntingtonovou nemocí a dlouho netušila, zda mutaci sama nese. Seriál poměrně realisticky ukázal, jak obtížné je rozhodování o prediktivním testování u zdravého člověka, protože nejde jen o lékařskou informaci, ale o znalost, která může zásadně změnit pohled na budoucnost, vztahy, plánování rodiny i smysl života.
Závěrem
Huntingtonova nemoc tak ukazuje, že dědičnost není vždy otázkou „máme určitý gen nebo ne“. Někdy totiž rozhoduje míra, do jaké je gen změněný. A první nemocný v rodině tak není ten, u koho se změna objevila, ale ten, u koho poprvé dosáhla kritické velikosti.
Na tomto místě je dobré dodat, že ačkoli nemoc zatím neumíme vyléčit, v loňském roce se díky genové terapii podařilo poprvé významně zpomalit její průběh, což je obrovský milník pro budoucí léčbu.
Pokud se vám moje příspěvky líbí, budu rád za sledování také na Instagramu.
Zdroje
Schultz JL, Moser AD, Nopoulos PC. The Association between CAG Repeat Length and Age of Onset of Juvenile‑Onset Huntington's Disease. Brain Sciences. 2020;10(9):575. doi:10.3390/brainsci10090575
Roudenský P. Genetic factors influencing age of onset of Huntington’s disease and markers of its prediction. Neurologie pro praxi. 2023;24(2):127-131. doi:https://doi.org/10.36290/neu.2022.064
Todd TW, Lim J. Aggregation formation in the polyglutamine diseases: Protection at a cost? Molecules and Cells. 2013;36(3):185-194. doi:10.1007/s10059-013-0167-x