


Článek
Protože jde o poměrně vzácné onemocnění, (u nás zhruba 200 nových případů ročně), většina lidí se s ALS setká až prostřednictvím konkrétních lidských osudů. Mnozí si vybaví fotbalistu Mariána Čišovského, bývalého premiéra Stanislava Grosse, dabéra Martina Kolára či překladatelku Danu Gálovou, které nemoc postihla.
ALS je onemocnění, kterému jsem se věnoval už během studií. Přiblížit jeho podstatu srozumitelně a lidsky mi přijde důležité, protože každý krok, který pomůže lidem nemoc pochopit, může přispět k většímu zájmu o výzkum a ke kvalitnější podpoře pacientů i jejich blízkých.
Co je vlastně ALS?
Název amyotrofická laterální skleróza zní složitě a většině lidí sám o sobě mnoho neřekne. Když ho ale rozebereme, pomůže pochopit, co se při této nemoci v těle odehrává.
Slovo amyotrofická označuje úbytek svalové hmoty, laterální odkazuje na postižení postranních oblastí míchy (viz obrázek níže), kde probíhají nervové dráhy přenášející signály z mozku do svalů, a skleróza znamená ztvrdnutí, které vzniká v místě odumřelých buněk. Kdybychom název volně vysvětlili, pak jde o nemoc, při které dochází k postupnému slábnutí a úbytku svalů v důsledku poškození a následného ztvrdnutí (sklerózy) nervových drah v postranních částech míchy, které tak ztrácejí funkci.
Zde je třeba uvést, že ačkoli název odkazuje na postižení míchy, ALS zasahuje i mozek, konkrétně mozkovou kůru, která vysílá povely k pohybu. Nemoc tak postupně narušuje celý motorický systém a schopnost ovládat svaly.
Toto narušení se konkrétně týká specializovaných nervových buněk, které pohybové povely přenášejí – motorických neuronů, neboli motoneuronů. Jsou to vlastně „vodiče“ pohybových signálů, uspořádané do dvou na sebe navazujících článků, jak ukazuje následující obrázek.
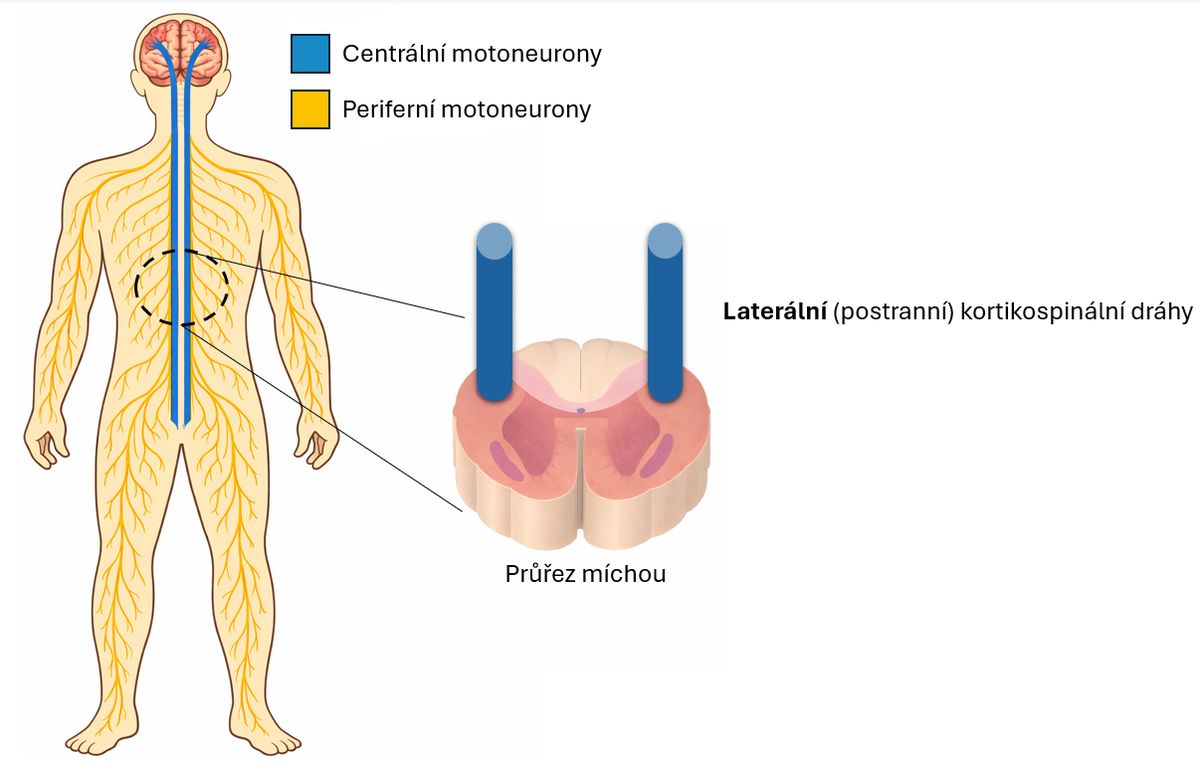
Zjednodušené schéma centrálního a periferního motorického neuronu. V průřezu míchou jsou zvýrazněny laterální kortikospinální dráhy, jejichž postižení se odráží v názvu onemocnění ALS – „laterální skleróza“.
První článek tvoří centrální motoneurony, které vysílají povely z mozku do míchy. Druhý článek, periferní motoneurony, pak tyto povely z míchy přenáší přímo do svalu.
Některé nemoci zasahují pouze centrální motoneurony, jiné postihují výhradně periferní motoneurony a projevy se pak liší podle toho, který článek nefunguje správně. U ALS jsou však postiženy oba články současně, a proto se objevují příznaky vycházející jak z centrálního, tak i z periferního motoneuronu.
ALS je postupně se zhoršující onemocnění, které medicína nedokáže vyléčit. Léčba je podpůrná a zaměřená na zmírnění příznaků a zajištění co nejlepší kvality života. Průměrná délka dožití od rozpoznání nemoci je obvykle 2–5 let, a ačkoli výjimky s výrazněji delším přežitím existují, jsou bohužel velmi vzácné.
Jak se ALS projevuje?
Onemocnění začíná nenápadně, skoro plíživě. První projevy lidé mnohdy dlouho přehlížejí, vysvětlují únavou, stresem nebo je přičítají přetížení, potížím s páteří či „skřípnutému nervu“.
V každodenních činnostech se začnou objevovat nenápadné potíže. Ruka je méně obratná, drobné úkony se zpomalují a vyžadují víc soustředění než dřív - typické je to u zapínání knoflíků, otáčení klíčem v zámku, otevírání sklenic a podobně. Při chůzi občas špička boty zavadí o práh, koberec nebo drobnou nerovnost, někdy je potřeba věnovat víc pozornosti při zvedání nohy přes obrubník nebo při nastupování do autobusu.
U části pacientů onemocnění začíná v oblasti hlavy a krku. Prvním příznakem je často postupné zhoršování výslovnosti, řeč se stává hůře srozumitelnou, a zejména delší mluvení stojí člověka více soustředění. Brzy se přidávají problémy s polykáním, nejprve jako občasné zakuckání po napití, ke kterému dochází čím dál častěji.
Ve svalech se prakticky u všech pacientů objevují drobné trhavé záškuby, známé jako fascikulace, a mnohdy také silné křeče.
Postupem času potíže sílí a šíří se do dalších svalových skupin. Slábnou nejen ruce a nohy, ale i svaly trupu, ramenního pletence a pánve, což ztěžuje běžné pohyby, stání i sezení. U pacientů s počátkem v oblasti hlavy a krku se řeč a polykání dále zhoršují a přibývá obtíží při každodenní komunikaci a jídle. Narůstající slabost postupně postihuje všechny svaly, včetně dýchacích a polykacích, kdy pacient ztrácí schopnost samostatně dýchat a jíst. Závislost na pomoci při všech běžných činnostech je kompletní, přičemž dlouhodobě zůstává zachována pouze hybnost očních svalů, která umožňuje jen omezenou komunikaci.
Kdo je ohrožen nejvíce?
Přibližně 5–10 % případů tvoří tzv. familiární ALS, kdy je onemocnění způsobeno konkrétními genetickými mutacemi, které se mohou v rodinách dědit z generace na generaci. Mezi nejlépe popsané patří mutace v genech SOD1, C9orf72, TARDBP a FUS, které zasahují do základních buněčných procesů, jako je zpracování bílkovin, práce s RNA nebo ochrana buněk před oxidačním stresem.
U těchto rodinných forem je riziko onemocnění výrazně vyšší, ale ani zde genetická mutace neznamená automaticky, že se ALS u daného člověka nutně rozvine. Průběh i věk nástupu se mohou lišit i mezi členy jedné rodiny, což ukazuje, že genetická predispozice sama o sobě nestačí a významnou roli hrají i další faktory.
Naprostá většina případů ALS je však tzv. sporadická, tedy vznikající bez známé rodinné zátěže. U těchto forem se předpokládá kombinace jemné genetické náchylnosti a vlivů prostředí, které společně překročí určitou biologickou hranici. Tyto faktory mohou zahrnovat dlouhodobý oxidační stres, poruchy buněčného metabolismu, chronický zánět nebo narušené mechanismy opravy nervových buněk. V posledních letech se intenzivně zkoumá role střevního mikrobiomu (blíže jsem psal zde), imunitního systému a celoživotních expozic různým toxickým látkám.
Riziko ALS roste s věkem, nejčastěji se onemocnění objevuje mezi 50. a 70. rokem života, i když výjimky existují a bohužel se lze setkat s pacienty výrazně mladšími i staršími. Z epidemiologických studií vyplývá, že muži onemocní o něco častěji než ženy, zejména v mladším a středním věku. Tento rozdíl se s narůstajícím věkem postupně stírá, což naznačuje, že ochrannou roli mohou hrát hormonální či metabolické faktory.
Možné spouštěče a rizikové faktory
U profesionálních sportovců, zejména hráčů amerického fotbalu, byla ALS zaznamenána až čtyřikrát častěji než u běžné populace a riziko stoupalo s délkou kariéry ve vrcholovém sportu. Podobně některé studie ukázaly vyšší výskyt nemoci i u hráčů evropského fotbalu. Zvýšené riziko se předpokládá také u lidí vystavených opakovaným drobným úrazům (zejména hlavy) či mikrotraumatům svalů a nervů, ke kterým může docházet při kontaktních sportech nebo při intenzivní fyzické zátěži.
Další oblastí zkoumání je vliv antibiotik. Švédská populační studie ukázala, že lidé, kteří v letech před diagnózou opakovaně užívali antibiotika, měli o něco vyšší pravděpodobnost rozvoje ALS než ti, kteří je neužívali, a riziko bylo vyšší u těch, kteří je užívali častěji. Přesný mechanismus není znám, ale předpokládá se, že by mohlo hrát roli narušení střevní mikroflóry a související změny imunitních či metabolických funkcí.
Rovněž je zkoumána souvislost s dlouhodobým vystavením některým chemikáliím, například pesticidům, herbicidům, určitým průmyslovým rozpouštědlům nebo těžkým kovům, u kterých studie naznačují mírně vyšší riziko rozvoje ALS.
Je však důležité zdůraznit, že tyto faktory pravděpodobně nejsou přímou příčinou ALS. Slouží spíše k identifikaci skupin lidí, u kterých se nemoc častěji objevuje, a poskytují vodítka pro další výzkum toho, jak mohou určité okolnosti ovlivnit riziko vzniku onemocnění.
Zdroje
Chapman, Laura, et al. „Physical Activity as an Exogenous Risk Factor for Amyotrophic Lateral Sclerosis: A Review of the Evidence.“ Brain: A Journal of Neurology, vol. 146, no. 5, 15 Mar. 2023, p. awac470, pubmed.ncbi.nlm.nih.gov/36918362/, https://doi.org/10.1093/brain/awac470.
Chio, A. „Severely Increased Risk of Amyotrophic Lateral Sclerosis among Italian Professional Football Players.“ Brain, vol. 128, no. 3, 5 Jan. 2005, pp. 472–476, https://doi.org/10.1093/brain/awh373.
Mead, Richard J., et al. „Amyotrophic Lateral Sclerosis: A Neurodegenerative Disorder Poised for Successful Therapeutic Translation.“ Nature Reviews Drug Discovery, vol. 22, no. 22, 21 Dec. 2022, https://doi.org/10.1038/s41573-022-00612-2.
Sun, Jiangwei, et al. „Antibiotics Use and Risk of Amyotrophic Lateral Sclerosis in Sweden.“ European Journal of Neurology, vol. 26, no. 11, 7 June 2019, pp. 1355–1361, https://doi.org/10.1111/ene.13986. Accessed 2 May 2023.






